 |
Center for Nonlinear Studies |
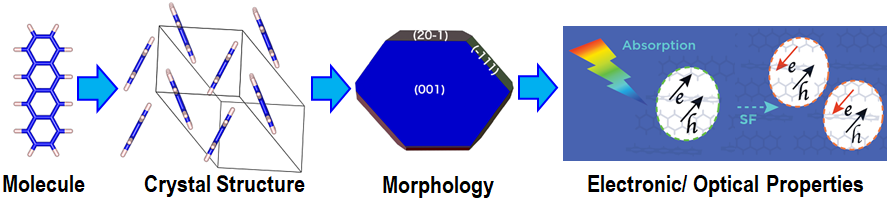
Molecular crystals are bound by dispersion (van der Waals) interactions, whose weak nature gives rise to polymorphism, the ability of a compound to crystallize in different structures. Crystal structure profoundly influences the physical and chemical properties, and hence the functionality of molecular solids in applications including pharmaceuticals, electronic devices, and energetic materials. Therefore, the ability to predict the structure and properties of molecular crystals is of paramount importance. To this end, we combine first principles simulations with machine learning.
Molecular crystal structure prediction (CSP) is challenging because it requires searching a high-dimensional configuration space with high accuracy. CSP workflow have two main components, structure generation and stability ranking. For structure generation, we develop the Genarris code [1], which generates random structures in all compatible space groups with physical constraints on intermolecular distances. Machine learned interatomic potentials (MLIPs) are trained on large data sets of first principles simulations [2], typically density functional theory (DFT) to achieve DFT-level accuracy at the computational cost of classical force fields. We have interfaced Genarris with several types of MLIPs for geometry optimization and stability ranking [1,3,4]. We have shown that MLIPs can completely replace both early-stage screening with classical force fields and final ranking with DFT, paving the way to high-throughput CSP [4].
One of the optoelectronic applications of molecular crystals is singlet fission (SF), the conversion of one photogenerated singlet exciton into two triplet excitons. SF has the potential to increase the efficiency of solar cells by harvesting two charge carriers from one high-energy photon, whose excess energy would otherwise be lost to heat. The realization of SF-based solar cells is hindered by the dearth of suitable materials. The excited-state properties of molecular crystals can be calculated using many-body perturbation theory (MBPT) within in the GW approximation and the Bethe-Salpeter equation (BSE) [5]. The computational cost of GW+BSE is prohibitive for large-scale exploration of the chemical space, and also for generating large amounts of training data. This calls for ML approaches that work well with small data. We have generated the PAH101 dataset [6] of GW+BSE results for 101 polycyclic aromatic hydrocarbons (PAH) and used the sure-independence-screening-and-sparsifying-operator (SISSO) ML algorithm to train models to predict the excited-state properties of crystalline organic semiconductors [6,7,8]. These models can be used to explore the chemical space and discover new candidate materials for organic optoelectronic devices.

[1] Y. Yang et al. ChemRxiv DOI: 10.26434/chemrxiv-2025-046zn (2025)
[2] V. Gharakhanyan et al., arXiv 2508.02651 (2025)
[3] K. S. Nayal et al. ChemRxiv DOI: 10.26434/chemrxiv-2025-ksn4n (2025)
[4] V. Gharakhanyan et al., arXiv 2508.02641 (2025)
[5] X. Wang et al. J. Phys. Chem. C 128, 7841 (2024)
[6] S. Gao et al. Scientific Data 12, 679 (2025)
[7] X. Liu et al., npj Comput. Mater., 8, 70 (2022)
[8] S. Gao et al. Digital Discovery 4, 1306 (2025)
Host: Nicholas Lubbers