| Geoffrey West |
Networks: a Matter of Life and Death? |
|
|
BANQUETT SPEAKER |
||
PROGRAM
Sunday, May 11, Pre-registration reception (food), 3:00 - 8:00 pm
|
Monday |
Tuesday May 13 |
Wednesday |
Thursday |
Friday |
|
| 8:15 - 9:00 |
8:45 - Meyer, Opening |
||||
| 9:00 - 9:45 | BRODER | KERTÉSZ | ALON | ||
| 9:45 - 10:00 |
COFFEE BREAK |
||||
| 10:00 - 10:45 | SETH | ALBERT | DAVIDSON, Bolouri | VIDAL | |
| 10:45 - 11:30 | VICSEK | KRAPIVSKY | SIGGIA | MENDES | |
| 11:30 - 12:15 | HUNTER | AUBOUY | CHANGEUX | REDNER | |
| 12:15 - 2:00 |
LUNCH |
||||
| 2:00 - 2:45 | HAVLIN | KLEINBERG | SOLÉ | KAHNG | |
| 2:45 - 3:30 | TREWHELLA | ALDOUS | MACKINTOSH | Socolar | |
| ben-Avraham | |||||
| 3:30 - 3:45 |
COFFEE BREAK |
||||
| 3:45 - 4:30 | ADAMIC | Eubank | OLTVAI | MONASSON | |
| Hastings | |||||
| Outlook - 4:45pm | |||||
| 4:30 - 5:15 |
KOHN, |
CALDARELLI | CHESWICK | NEWMAN | |
| 5:15 - 6:15 |
Cash Bar |
||||
| 6:15 -- 7:00 |
Reception, |
Posters |
Posters |
||
| 7:00 -- 9:00 |
Posters |
Posters |
BANQUET |
Posters | |
|
|
|
||||
|
|
|||||
EVERY
INVITED SPEAKER HAS 30
minutes RESERVED
FOR HER/HIS TALK
and
15 minutes
RESERVED FOR QUESTIONS
Short talks are 12 minutes
plus 10 minutes questions
(short talks were introduced after some
cancellations, and they were chosen in the order of original requests.
No more short talks can be inserted at this time)
| Geoffrey West |
Networks: a Matter of Life and Death? |
|
|
BANQUETT SPEAKER |
||
| Lada Adamic |
Information flow in social groups |
|
| We present a study of information flow that takes into account the observation that an item relevant to one person is more likely to be of interest to their immediate neighbors than to those multiple steps removed. This is due to the fact that the similarity of node attributes in social networks decreases as a function of the graph distance. An epidemic model on a scale-free network with this property has a finite threshold, implying that the spread of information is limited. We have tested our predictions by measuring the propagation distance of messages in an organization and also by numerical experiments that take into consideration the organizational distance among individuals. | ||
| Réka Albert |
Topology and function of the segment polarity gene network |
|
| Biological systems often form complex networks of interaction. For example, the expression of genes is regulated by interactions with other genes and gene products. Recent experimental advances uncovered the qualitative structure of many gene control networks, creating a surge of interest in the quantitative description of gene regulation. In this talk I will focus on the segment polarity genes of the fruit fly Drosophila melanogaster, and the network of interactions determining the stability of their expression. I will present a Boolean representation of this network that assumes that genes and proteins are either ON or OFF, and their interactions can be formulated as logical functions. This model is able to reproduce the observed spatial patterns of the segment polarity genes as well as the patterns obtained in gene mutation experiments. In addition, the model gives insights into the functioning of the network, suggesting a remarkable robustness towards changes in internal parameters, initial conditions and even some mutations. | ||
| David Aldous |
A Tractable Complex Network Model Based on the Stochastic Mean-Field Model of Distance |
|
| Much recent research activity has been devoted to empirical study and theoretical models of complex networks possessing three qualitative features: power-law degree distributions, local clustering, and slowly-growing diameter. We point out a new (in this context) platform for such models -- the stochastic mean-field model of distances -- and within this platform study a simple two-parameter proportional attachment model. The model is mathematically natural, permits a wide variety of explicit calculations, has the desired three qualitative features, and fits a broad range of degree scaling exponents and clustering parameters; in these respects it compares favorably with existing models. | ||
| Uri Alon |
Network motifs: simple building blocks of complex networks |
|
| Complex networks of interactions are studied across science. For example, a major goal of systems biology is to understand the dynamics of regulatory networks that control cell responses to external signals. In order to break networks down into basic building blocks we defined 'network motifs': patterns of connections that recur in the network much more often than in randomized networks. We find that the transcription networks of E. coli and yeast are composed of several highly significant network motifs. We analyzed these motifs experimentally by means of high-resolution gene expression experiments with green fluorescent protein reporter libraries developed in our lab. We find that each of the network motifs has a specific information processing role, such as filtering fluctuations in input stimuli or generating temporal patterns of expression. We demonstrate how this approach can be used to detect motifs in other biological networks, such as the synaptic connection neuronal network of C. elegans. Some of the network motifs in the neuronal network are the same as the network motifs found in transcription networks, raising the possibility that these shared circuit patterns perform similar information processing tasks. The network motif detection algorithms can be applied to define the building blocks of virtually any network, as we demonstrate using technological and ecological network databases. | ||
| Miguel Aubouy |
A strain-like variable to quantify the deformation of connected networks. |
|
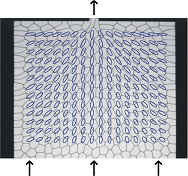 |
Connected networks are networks of sites connected by links, which can detach from and reattach to other sites. Cellular patterns can be described as connected networks if sites are the meeting points of cells, and two sites connects if their cells share and edge. There is a variety of issues where we need to quantify the deformation of connected networks. For example, when we have to measure the anisotropy of cellular patterns, or when we analyse the flow of two dimensional liquid foams. In this talk, we discuss these example in some details. We introduce a so-called texture tensor to characterize the structure of the material. In particular, we show that image analysis of inhomogeneous flow of 2d foams (Cf Figure) yields informations on the elastic constants of this material. | |
| Joint work with: François Graner, Yi Jiang, James Glazier, Benjamin Dollet, Marius Asipauskas | ||
| Robert Axtell |
Job Search Networks |
|
|
In an individual agent model of firm formation and
dynamics, the role of inter-firm migration--job-changing--is
investigated. Agents change jobs on the basis of information gleaned
from their social networks. The character of these networks alters the
stationary distribution of firm sizes that arises in the model. We infer
the structure of such networks by calibrating model output to the
empirical distribution of firm sizes in the U.S.
|
||
| Albert-László Barabási |
The architecture of complexity: From the topology of the www to the cell's genetic network |
|
| Networks with complex topology describe systems as diverse as the cell or the World Wide Web. The emergence of these networks is driven by self-organizing processes that are governed by simple but generic laws. The analysis of the metabolic and protein network of various organisms shows that cells and complex man-made networks, such as the Internet or the world wide web, share the same large-scale topology. I will show that the scale-free topology of these complex webs have important consequences on their robustness against failures and attacks, with implications on drug design, the Internet's ability to survive attacks and failures, and our ability to understand the functional role of genes in model organisms. | ||
| Daniel ben-Avraham |
Random Walks on Scale-Free Networks |
|
| Random walks serve as a pedestrian model for the propagation of information in computer networks, rumors, or disease in social networks, etc. We study the speed of propagation by examining the first passage time (FTP) of random walkers on scale-free networks. We focus on deterministic, scale-free, recursive networks, where we can compute the FTP between any two nodes analytically. Some preliminary conclusions as to which topological aspects inhibit or aid propagation follow from comparisons between scale-free networks with and without cycles (loops), as well as to Complete graphs and Cayley trees. | ||
| Andrei Z. Broder |
Practical challenges in the study of the web-graph |
|
| The most reliable way to
discover the hypertext graph structure of the world wide web is via large
scale crawls. However, the picture that emerges is still subject to
distortions induced by the size and speed limitations of the crawl, by
various crawling and representation policies, and by
"legitimate" and "spam" perturbations of the
"natural" process of birth and death of nodes and links.
Furthermore, given the size of the web-graph, representing and exploring
it is far from trivial. This talk will survey these phenomena, illustrate them with real life examples, and discuss certain techniques that could be used to alleviate some of these issues. |
||
| Guido Caldarelli |
Networks structures in Financial Markets |
|
| We are going to present the evidence of some networks structures in financial markets. We will focus on the correlation between price returns in different stocks and we also considere a different structure represented by the network of shareholders in a market. IN the first case the topological properties of the market can mainly be used in order to assess the validity of various market models. In the second case, the topological structure can also help in detecting the behaviour of different markets. Furthermore in the latter case, we believe that the evidence of fat tails in the degree distribution might be related to a generalized Pareto law. | ||
| Jean-Pierre Changeux |
A MODEL OF CONSCIOUS WORKSPACE INVESTIGATED WITH COGNITIVE TASKS AND KNOCK-OUT MICE |
|
| A minimal
hypothesis is proposed concerning the brain processes underlying effortful
tasks. It distinguishes two main computational spaces: a unique global
workspace composed of distributed and heavily interconnected neurons with
long-range axons, and a set of specialized and modular perceptual, motor,
memory, evaluative, and attentional processors. Workspace neurons are
mobilized in effortful tasks for which the specialized processors do not
suffice. They selectively mobilize or suppress, through descending
connections, the contribution of specific processor neurons. In the course
of task performance, workspace neurons become spontaneously coactivated,
forming discrete though variable spatio-temporal patterns subject to
modulation by vigilance signals and to selection by reward signals. A
computer simulation of the Stroop task shows workspace activation to
increase during acquisition of a novel task, effortful execution, and
after errors. Predictions for spatio-temporal activation patterns during
brain imaging are outlined particularly the contribution of dorsolateral
prefrontal cortex and anterior cingulate to the workspace. Pharmacological
and pathological implications in humans are underlined, and possible tests
with genetically modified mice examined. |
||
| William R. Cheswick |
Mapping the Internet and Internets |
|
| Hal Burch and I started the Internet Mapping Project in the summer of 1998 to try to collect long-term, consistant topological information about the "center" of the Internet. We record the output of hundreds of thousands of traceroutes daily, and have accumulated a database of Internet paths. The techniques, and others, have been refined to examine intranets for industry and the US government, and other networks of political interest. We also developed spring-force simulation graph-layout algorithms to display network graphs of much larger sizes than were thought feasible at the time. | ||
| Fan Chung Graham |
Spectra of random graphs with given expected degrees |
|
| In the study of the spectra of power law graphs, there are basically two competing approaches. One is to prove analogues of Wigner's semi-circle law while the other predicts that the eigenvalues follow a power law distributions. Although the semi-circle law and the power law have nothing in common, we will show that both approaches are essentially correct if one considers the appropriate matrices. We will prove that (under certain mild conditions) the eigenvalues of the (normalized) Laplacian of a random power law graph follow the semi-circle law while the spectrum of the adjacency matrix of a power law graph obeys the power law. Our results are based on the analysis of random graphs with given expected degrees and their relations to several key invariants. Of interest are a number of (new) values for the exponent b where phase transitions for eigenvalue distributions occur. The spectrum distributions have direct implications to numerous graph algorithms such as randomized algorithms that involve rapidly mixing Markov chains, for example. | ||
| Joint work with: Lincoln Lu and Van Vu | ||
| Eric Davidson |
The endomesoderm network, a logic map for early development: Tests of cis-regulatory predictions |
|
| The regulatory program for animal development is "wired" into the cis-regulatory networks of the genome. At the DNA level these networks consist of the clusters of cis-regulatory transcription factor target sites (modules) that direct the spatial and temporal expression of each phase of activity of those genes; and the linkages amongst them. Here "linkage" refers to the relation between genes encoding transcription factors and the target sites of genes which they control, and between the genes encoding elements of signal systems and their ultimate target genes. We are engaged in an effort to define at the genomic sequence level the gene regulatory network (GRN) controlling endomesoderm specification in sea urchin embryos. The proposed GRN is based on the following information: spatial and temporal expression patterns of all genes included; constraints from experimental embryology; and a massive perturbation analysis in which expression of every relevant gene is perturbed and the effects on all other genes measured. The GRN consists of predicted inputs into the cis-regulatory modules controlling the endomesoderm expression of the genes involved. These predictions can be tested at the cis-regulatory level: here several such tests are presented. They show that the perturbation analysis is a surprisingly informative predictor of DNA sequence-level regulatory transactions. | ||
| John Doyle |
HOT FAST Networks |
|
| A widely shared vision is that there are universal features of complex systems that transcend reductionist decompositions, but there are sharp differences regarding what those features are. This talk is inspired by the striking convergence of three research directions. First, biologists have provided a detailed description of the components of biological networks, and many organizational principles of these networks are becoming increasingly apparent. Even bacterial cells integrate communication, computation, and feedback control subsystems into highly organized regulatory networks. Second, advanced information technologies are approaching biology in their complexity. While the components are entirely different, there are remarkable similarities at the network architecture level and in the role of protocols in structuring modularity, with layers of feedback and regulation. New theories elucidate these similarities. We have the beginnings of the first coherent and complete theoretical foundation of the Internet, which has led to successful large scale test deployments of new protocols, and have also been developing new theory and software infrastructure to support systems biology. Finally, and most importantly, a new mathematical framework makes rigorous and precise the notion that this apparent network-level evolutionary convergence within and between biology and technology is not accidental, but follows necessarily from the universal requirements of efficiency and robustness. It is in retrospect unsurprising that a genuinely new science of complexity would require equally new mathematics, in this case combinations of pure mathematics not traditionally thought of as applied. A key feature is that the ``robust yet fragile" nature of complex systems that has been a theme of HOT has the computational counterpart of ``dual complexity implies primal fragility." Organisms, ecosystems, and successful advanced technologies are highly constrained in that they are not evolved/designed arbitrarily, but necessarily in ways that are efficient yet robust to uncertainties in their environment and their component parts. These are extremely severe constraints, not present in other sciences but essential in both biology and engineering. Failure to explicitly exploit the highly structured, organized, and ``robust yet fragile" nature of such systems hopelessly dooms traditional methods to be overwhelmed by their sheer complexity. Though still very new, the new mathematics and software based on it have already found substantial applications not only to networking and biology but also to algorithms, hybrid and nonlinear control, multiscale physics, and finance. A side benefit of a deepening understanding of the fundamental nature of complexity in a general sense is also new and more rigorous explanations for long-standing problems in physics associated with complex systems, such a the nature and structure of shear flow turbulence, the ubiquity of power laws, the origin of dissipation and entropy, and quantum entanglement and measurement. | ||
| Stephen Eubank |
tba |
|
| Matthew Hastings |
Network Field Theory |
|
| We use scaling results to identify the crossover to mean-field behavior of equilibrium statistical mechanics models on a variant of the small world network. The results are generalizable to a wide-range of equilibrium systems. Anomalous scaling is found in the width of the mean-field region, as well as in the mean-field amplitudes. Finally, we consider a non-equilibrium processes: the contact process for disease spreading. | ||
| Shlomo Havlin |
Stability and tomography of complex networks |
|
| General results for complex networks, such as the distance, optimal paths, the tomography of the network (statistical properties of different layers) as well as an efficient strategy for immunization will be discussed. A percolation approach for the behavior of generalized random graphs with a known connectivity distribution of links k per node, P(k), under both random breakdown of nodes and intentional attack on the most highly connected nodes will be presented. Specifically, scale free networks, such as the Internet, where P(k) ~ k-a with a<3 are resilient to random breakdown, but very sensitive to intentional attack. | ||
| John Hopfield |
Network coordination and the 'atoms' of speech perception |
|
| While speech can be transcribed into phonemes, they are neither the elements of speech perception nor of speech generation. We describe a model network of spiking neurons based loosely on known properties of vertebrate auditory systems. The system spontaneously displays a rich set of 'atoms of speech perception' in the presence of a weak common input from a global coordinating rhythm that completely alters the computational potential of the network. | ||
| Tony Hunter |
Protein
phosphorylation and intracellular signaling networks in the postgenomic
era |
|
| Protein kinases and phosphatases are key components of protein phosphorylation based signaling networks in eukaryotic cells. In consequence, there has been long-standing interest in the number of protein kinases (PKs) and phosphatases (PPs) needed to constitute intracellular signaling networks. We have used complete eukaryotic genome sequences combined with cDNA and EST sequence information to determine the total number of PK genes in a given eukaryote (the kinome). The human kinome has 518 PK genes and 106 kinase-derived pseudogenes; 478 of these PKs belong to the eukaryotic PK (ePK) superfamily, and the remainder are atypical PKs (aPK), divided among a few small families. Over 50 of the ePK catalytic domains in the human kinome lack one of more of the key ePK catalytic residues, and are therefore predicted to lack catalytic activity; these domains presumably have functions other than phosphate transfer. Comparison of the kinomes from different eukaryotic species provides insights into the evolutionary history of the PKs. PKs constitute ~2% of all genes in budding and fission yeast, worms, flies and humans, and ~4% in plants. True tyrosine kinases (TKs) are lacking in single cell yeasts, but are present in simple metazoans, suggesting that tyrosine phosphorylation evolved as a mechanism for intercellular communication. In metazoans 15-20% of all PKs are TKs. Mutations in PK and PP genes are increasingly found to be causal in human diseases; 244 human PKs map to disease loci or to cancer amplicons, and ~20% of all oncogenes implicated in human cancer encode activated TKs. In consequence, PKs have become major drug targets. | ||
| Byungnam Kahng |
Classes in the shortest pathways of scale-free networks |
|
|
While the degree distributions give structural
information of complex networks, the degree exponent g
turns out to be sensitive to the details of network structures. Here we
study a problem of data packet transport between a pair of vertices in
scale-free networks. We introduce the load of a vertex as the
accumulated sum of data packets traveling along the shortest pathways
between pairs of vertices, which in statistical sense is the same as the
betweenness centrality (BC) often used in social networks. The load or
the BC distributions reflect the shortest pathway topologies imbedded in
them. It is found that the load or the BC distribution follows a
power law with an exponent d
which is either 2.2(1) (class I) or 2.0 (class II), insensitive to
different values of g
in the range, 2 < g
<(=) 3 and different mean degree. The classification of scale-free
networks into the two classes stems from the characteristics of the
shortest pathways of scale-free networks. While the shortest pathways
between a pair of vertices are multiplely connected in the class I, they
are almost solely connected in the class II. The prototypical examples
of the class I and II are the protein interaction network and the
Internet on the autonomous system level, respectively. Such distinct
topological features of the shortest pathways produce different
behaviors in diverse problems such as the resilience of networks, i.e.,
the diameter changes under the removal of a single vertex and in a
synchronization problem. We also discuss the load-load correlations in
assortative, neutral, and dissortative networks, and various other
problems in relation to shortest pathways.
|
||
| János Kertész |
Structure and properties of complex networks |
|
| We study the structural phase transition of scale free networks related to clustering. We present a scaling framework and a rate equation based mean field theory, the latter leading to the widely observed 1/k dependence of the clustering coefficient. We also investigate, mainly numerically, spreading phenomena on Barabási-Albert graphs. The weight distribution on minimum spanning trees is calculated and we observe for uniformly distributed random weights (1-w)m-1 for m -> 1. For invasion percolation the equivalence to ordinary percolation seems to brake down. | ||
| Co-Authors: Gabor Szabo and Mikko Alawa | ||
| Jon Kleinberg |
Information Flow and Gossip-Based Algorithms in Decentralized Networks |
|
| The dynamic behavior of a network in which information is changing continuously over time requires robust and efficient mechanisms for keeping nodes updated about new information. A promising approach to this problem is to employ highly decentralized algorithms in which information is spread by random node-to-node ``gossip''; in this way, new updates are propagated in epidemic fashion, and will reach most nodes despite failures and lack of coordination. We consider the basic structure of these algorithms, and the dynamics they induce on the network; we also focus on the particular problem of spreading information when geographic locality is an issue. We find that subtle changes in the underlying rules used for information-spreading -- particularly the balance between short-range and long-range communication -- can have qualitative global effects on the overall dynamics. | ||
| Co-Authors: David Kempe and Alan Demers | ||
| Kurt Kohn |
A Graphic Notation for Bioregulatory Networks |
|
| A vast amount of information has accumulated about the molecular interactions that govern cell behavior, such as cell division, differentiation, and death. Just as electronic devices need circuit diagrams, the equivalent is needed for bioregulatory networks in order to comprehend, trouble-shoot, or modify their behavior. This is especially important in designing pharmacological intervention for therapy. Diagramming the control networks that regulate cell functions however presents special difficulties, due to the prominent role of multimulecular complexes and protein modifications. A given molecule can exist in a large number of states, depending on other molecules to which it may be bound and on the modification states (such as phosphorylations) of these molecules. Often the molecules that add or remove these modifications are themselves targets of modification and complex formation. All this can give rise to an unmanageable "combinatorial explosion." Standard circuit diagrams or classical biochemical pathway diagrams cannot manage these complexities without creating an incomprehensible mess. We often have only partial knowledge of the multiple paths to the assembly of a multimolecular complex. Each molecule in a complex may have multiple modification states that affect function, and we usually have only partial information about these effects. The incompleteness of our knowledge presents problems of ambiguity and incompleteness that standard circuit diagrams or diagrams suitable for computer simulation cannot tolerate. We have therefore devised a notation that usually can cope with these problems. It allows us to depict the interactions and effects that we know, while leaving some of the details undefined or represented in an abbreviated form that covers multiple possibilities. One useful feature is that each monomolecular species is represented only once in a diagram. Hence, all the interactions involving a given molecular species can easily be traced. Each interaction is linked to an annotation that provides additional information and literature citations. Any molecule or interaction can be found by means of an index of coordinates, as in an ordinary roadmap. A set of symbols is defined for these "heuristic" diagrams. A subset of these symbols can be used to create unambiguous ("explicit") diagrams for computer simulation. (References: Kohn 1999 Molec. Biol. Cell 10: 2703-2734; 2001 Chaos 11: 84-97.) | ||
| György Korniss |
Controlling Rough Virtual Times via Small-World Synchronization |
|
| We study the scalability of parallel simulations for systems with non-parallel (asynchronous) dynamics. We look at the simulation itself as a complex evolving systems where the individuals are the processing elements (PE), which interact through a synchronization scheme. It was shown that under the basic conservative scheme for systems on regular lattices with short range interactions, the progress of the simulation is governed by the Kardar-Parisi-Zhang equation. While it ensures a finite average progress rate of the simulation, the spread (``width'') of the progress among the individual PEs diverges in the long-time, large number of PE limit. This latter property can hinder scalable data management. Considering a stochastic growth process on a quenched random (small-world-like) network, we construct a scheme where the PEs make a non-zero and close-to-uniform progress, making the simulation fully scalable. | ||
| Paul Krapivsky |
Growing Networks: Local and Global Characteristics |
|
| In this talk I will outline local and global characteristics of growing networks. I will show that the rate equation approach is a simple yet powerful tool to analyze growing network systems. I will show that the power-law in- and out-degree distributions are dynamically generated in a simple model that mimics the web graph. I will also discuss interesting infinite-order percolation transition that often occurs in growing networks. | ||
| Ying-Cheng Lai |
tba |
|
| Fred MacKintosh |
Elasticity and dynamics of cytoskeletal networks |
|
| Networks of filamentous proteins play a crucial role in cell mechanics. These cytoskeletal networks, together with various crosslinking and other associated proteins largely determine the (visco)elastic response of cells. The response of these networks is shown to be highly non-linear, and the shear moduli can vary by orders of magnitude with small changes in density and local network connectivity. We discuss recent models of the elastic and dynamics properties of these cytoskeletal networks. We find, among other things, a crossover from affine to non-affine response of these semi-flexible polymer networks. Interestingly, this crossover is completely separate from rigidity percolation in such systems, which we also discuss. We represent scaling arguments that capture the essence of this intriguing breakdown of continuum elasticity on mesoscopic length scales in the system. | ||
| José F.F. Mendes |
Spectral analysis of complex networks |
|
| Significant research efforts have recently been made in the analysis of networks topology. Also, several studies were proposed to study the density of states of random and scale-free graphs. I will present some recent studies about the spectra of the adjacency matrices of correlated and uncorrelated random Bethe lattices. Exact equations will be presented. Together, a simple approximation will be used to calculate the spectra of random and scale-free graphs. The results will be compared with previously obtained (numerical, simulations and empirical) results. | ||
| Rémi Monasson |
tba |
|
| Mark Newman |
Mixing patterns in networks |
|
| The patterns of connection between vertices in a network are by no means random, and in particular they are often influenced by the different types or properties of vertices. For example, connections in a social network are frequently between individuals who are similar in some way, such as having the same language, age, or race. This phenomenon is known as "assortative mixing". I will describe some recent empirical work on assortative mixing in networks, including both social networks and other network types, and also discuss a variety of analytic and numerical results that help us to understand the consequences of different mixing patterns for the behavior of networked systems. | ||
| Zoltán Oltvai |
Organization of cellular networks |
|
| One of the most important goals of post-genomic biology is the elucidation of the fundamental logic and constraints that determine the systemic behavior of a cell or microorganism. In their totality, the combined interaction of various cellular constituents forms a complex cellular network whose functional organization is an important determinant of all cellular behaviors. Our research aims to understand the organization of this network from various perspectives, and with a combination of experimental and theoretical approaches. In our recent work we have demonstrated that the large-scale organization of metabolic-, transcriptional- and protein interaction networks appear identical, all possessing a robust and error tolerant scale-free topology with hierarchically embedded modularity. Within E. coli the uncovered hierarchical modularity closely overlaps with known metabolic functions, suggesting that the identified network architecture may be generic to system-level cellular organization. The results of systematic experimental verification of this framework by global transposon mutagenesis and the evolutionary consequences of such organization will be discussed. | ||
| Christos Papadimitriou |
ON CERTAIN RANDOM GRAPH MODELS OF THE INTERNET |
|
| This talk surveys some recent mathematical results pertaining to the Internet. We present a model of network creation based on bi-criterion optimization that predicts heavy-tailed degrees, and another based on Nash equilibria of a simple game. We show that certain "scale-free" random graphs models have interesting properties (expansion, linear growth of congestion with size, small overpayment in shortest path auctions, largest eigenvalues obeying a power law). | ||
| Prabhakar Raghavan |
Social networks in knowledge management |
|
| Beginning with Milgram's classic "six degrees of separation" experiments in the 1960's, social network theory has evolved to study how individuals interact and disseminate information. More recently, the study has come to encompass the broader network involving people as well as the documents and information they interact with - examples include recommendation systems and the use of link analysis in web search engines. This talk will provide an overview of this evolving area and the many research problems that arise, including issues in privacy protection. | ||
| Sidney Redner |
Extremal Properties of Growing Networks |
|
| The rate equation approach is exploited to determine extremal characteristics of growing networks. We first determine the joint age-degree distribution of such networks and show how "old" nodes typically have a large degree. We develop this idea to determine statistical properties of the nodes with the highest degree. We thus show that the number of lead changes among the nodes with the highest degree increases logarithmically with network size N, independent of the details in the growth mechanism. The probability that the first node retains the lead approaches a finite constant for popularity- driven growth, and, more interestingly, decays as N-f (ln N)-1/2, with f=0.08607...., for growth with no popularity bias. | ||
| Co-Authors: P.L. Krapivsky | ||
| Anil K. Seth |
Theoretical neuroanatomy:
Analyzing the structure and dynamics of networks in the brain |
|
|
Anatomically, the brain can be described as a highly non-random network of neurons. While recent developments in graph theory can help characterize the structural properties of this network, a fundamental challenge for neuroscience is to understand how such structure relates to neural dynamics. We have combined graph theory with aspects of statistical information theory to analyze the relations between structure and dynamics in simple computational models of cortical neural networks [1] . Specifically, we describe a quantitative measure of the ‘complexity’ of network dynamics, high values of which reflect an even balance between functional integration (large subsets tend to behave coherently) and functional segregation (small subsets tend to behave independently). Using an evolutionary graph-selection procedure, we found that networks with highly complex dynamics had structural motifs characteristic of the cerebral cortex - dense groups of highly interconnected nodes linked by a relatively small number of reciprocal bridges. Furthermore, known connectivity matrices of cat cortex and macaque visual cortex turn out to be near-optimal for generating complex dynamics. We have suggested that complex dynamics may provide robust and adaptive responses when networks are transforming input signals into output signals. To test this suggestion, we evolved networks to control target fixation in a simulated head/eye system, comparing the dynamical properties of networks evolved in simple and challenging task conditions. Only those networks which were evolved in challenging conditions displayed robust and adaptive behaviors; these networks also showed comparatively high dynamical complexity. These findings are unlikely to be limited to theoretical neuroanatomy: complex dynamics may underlie adaptive responses in networks of many different kinds. This research was made possible by the Neurosciences Research Foundation, which supports the work of The Neurosciences Institute. |
||
| Co-Author: Gerald M. Edelman | ||
| Eric D. Siggia |
Gene regulation during early development of the fly |
|
| The genome contains both information about the 'parts' (eg proteins) of the organism as well as the instructions to assemble it, encapsulated into 'cis regulatory modules'. The generation of new forms through evolution proceeds as much by regulatory innovation as by the generation of novel proteins. Development is a particularly striking instance of how a cascade of regulatory factors leads to the formation of a body plan from the egg. For the fly, I will show how compuational methods can extract some the regulatory code used in early development from the genome, illustrate these predictions with experiments, and provide some preliminary data on how these regulatory interactions evolve. | ||
| Joshua Socolar |
Scaling in Random Boolean Networks |
|
| We analyze the properties of random Boolean networks in the ordered regime and at the critical point. In the ordered regime, the structure of the networks simplifies in the large N limit. The average number of nodes, r, that determine the attractor dynamics remains constant with increasing system size and the nodes are organized into trivial dynamical loops. At the critical point r goes like N^(1/3). Numerical experiments also show that the median number of attractors in critical networks grows faster than linearly with N. | ||
| Co-Authors: Stuart Kauffman | ||
| Ricard V. Solé |
SELECTION AND EMERGENCE IN COMPLEX NETWORKS |
|
| Complex biological networks have very different origins than technologic ones. The latter involve extensive design and, as engineered structures, include a high level of optimization. The former involve (in principle) contingency and structural constraints, with new structures being incorporated through tinkering with previously evolved modules or units. However, the observation of the topological features of different biological nets suggests that nature can have a limited repertoire of "attractors" that essentially optimize communication under some basic constraints of cost and architecture or that allow the biological nets to reach a high degree of homeostasis. Conversely, the topological features exhibited by some technology graphs indicate that tinkering and internal constraints play a key role, in spite of the "designed" nature of these structures. Previous scenarios suggested to explain the overall trends of evolution are re-analyzed in light of topological patterns. | ||
| Eugene H. Stanley |
Optimal Paths in Disordered Complex Networks |
|
| We study the optimal distance lopt in small-world and scale-free networks in the presence of disorder implemented by assigning random weights to the links. We define lopt as the length of the path that minimizes the sum of the weights along the path. For strong disorder, where the maximal weight along the path dominates the sum, we find that lopt ~ N1/3 in both ER and WS networks. We also find that for scale-free networks with degree distribution P(k) ~ k-l, lopt scales as N(l-3)/(l-1) for 3 < l< 4 and as N1/3 for l >(=) 4. For 2 < l < 3, our numerical results suggest that lopt scales as logl-1N. We show that for weak disorder lopt ~ log N for both the Erdõs-Rényi (ER) and Watts-Strogatz (WS) models as well as for scale-free networks. | ||
| Co-Authors: Lidia Braunstein, Sergey Buldyrev, Reuven Cohen, and Shlomo Havlin | ||
| Jill Trewhella |
Intracellular Signaling: where experiment meets modeling and the data are scarce! |
|
| As we aspire to achieving a systems level understanding of a living cell, we must grapple with the challenge of thinking about thousands of macro-molecules undergoing conformational transitions as well as dynamic fluctuations as they form sometimes transient interactions in going about their respective functions. This symphony of movement and chemistry is highly regulated, each event carefully orchestrated by a set of signals. In the world of molecular structural biology, we are rapidly generating information on component structures (or subunits) of biological systems while driving toward understanding how they interact and form functioning assemblies. I will describe how modeling and experiment combine to provide insights into intracellular signaling networks that are controlled via second messengers, such as divalent calcium of cyclic nucleotides, and by protein phosphorylation. | ||
| Alessandro Vespignani |
Epidemic spreading in complex networks |
|
| We present a review of recent results about the spreading of infections in complex population networks. We show that the large connectivity fluctuations usually found in these networks strengthen considerably the incidence of epidemic outbreaks. Scale-free networks, which are characterized by diverging connectivity fluctuations, exhibit the lack of an epidemic threshold and always show a finite fraction of infected individuals. This particular weakness is found also in immunization strategies that can be successfully developed only by taking into account the inhomogeneous connectivity properties of scale-free networks. The understanding of epidemics in complex networks might deliver new insights in the spread of information and diseases in many biological and technological networks that appear to be characterized by complex heterogeneous architectures. | ||
| Tamás Vicsek |
Topological phase transitions |
|
| To provide a phenomenological theory for the microscopic dynamics of the restructuring in networks we use a statitical mechanics based approach with detailed balance satisfied for the transition probabilities between topological states. This enables us to establish the equivalence of our graph rewiring problem with the dynamics in a lattice gas on a nearly fully connected network. By defining the appropriate order parameters we find a rich variety a topological phase transitions (singular changes in the degree distribution as a function of a parameter playing the role of the temperature). In the "critical point" scale free networks are recovered. | ||
| Co-Authors: I. Derenyi, I. Farkas, G. Palla | ||
| Marc Vidal |
Integrating “omic” information: a bridge between genomics and systems biology |
|
|
Despite
the considerable success of molecular biology to understand diseases such
as cancer, many fundamental questions remain unanswered. Most importantly,
since the majority of gene products in the cell mediate their function
together with other gene products, biological processes should be
considered as complex networks of interconnected components. In other
words, for any normal biological process, or any disease mechanism, such
as cancer, one might consider a “systems approach” in which the
behavior and function of such networks are studied as a whole, in addition
to studying some of its components individually. The draft of the human
genome sequence is likely to help such a transition from molecular biology
to systems biology. Our
laboratory uses a model organism, the nematode C. elegans, to study the role of protein networks in development
and, doing so, develop the concepts and technologies needed for a
transition to systems biology. Our goals are to: i)
generate protein-protein interaction, or 'interactome', maps for C.
elegans networks involved in development, ii)
develop new concepts to integrate such interactome maps with other
functional maps such as expression profiles (transcriptome), global
phenotypic analysis (phenome), localization of expression projects (localizome),
etc…. and iii)
use such integrated information to discover novel network properties. |
||
| Andreas Wagner |
The design of large genetic networks: function, history, or (mere) chemistry? |
|
| Functional genomics is generating much qualitative information about the structure of genetic networks. How much biology can we learn from such qualitative information? I will address this question in the context of the yeast protein interaction network and the E. coli metabolic network. Specifically, I will ask whether these networks have their observed structure because this structure provides robustness against mutations. I will also ask whether this structure contains information about the evolutionary history of these networks. | ||
| Peter Wolynes |
Stochastic gene expression as a many-body problem |
|
| Gene expression has a stochastic component because of the single-molecule nature of the gene and the small number of copies of individual DNA-binding proteins in the cell. We show how the statistics of such systems can be mapped onto quantum many-body problems. The dynamics of a single gene switch resembles the spin-boson model of a two-site polaron or an electron transfer reaction. Networks of switches can be approximately described as quantum spin systems by using an appropriate variational principle. In this way, the concept of frustration for magnetic systems can be taken over into gene networks. The landscape of stable attractors depends on the degree and style of frustration, much as for neural networks. We show the number of attractors, which may represent cell types, is much smaller for appropriately designed weakly frustrated stochastic networks than for randomly connected networks. | ||